再生医療等製品の開発に携わる皆様、日々革新的な治療法の実現に向けてご尽力されていることと存じます。
特に治験プロトコルの策定段階では、「希少疾患で症例が集まらない」「外科的処置を伴うためプラセボ対照が難しい」といった、再生医療特有の壁に直面されている方も多いのではないでしょうか。
従来の医薬品開発における「ゴールドスタンダード」であるランダム化比較試験(RCT)が実施困難な場合、どのように科学的妥当性を担保し、規制当局の承認を得ればよいのか、頭を悩ませる場面も少なくありません。
本記事では、再生医療における治験デザインの考え方から、具体的な試験デザインの種類、PMDA(医薬品医療機器総合機構)との相談をスムーズに進めるポイントまでを詳しく解説いたします。
規制の枠組みを正しく理解し、製品の特性に合わせた柔軟かつ戦略的な治験デザインを設計するためのヒントとして、ぜひお役立てください。
再生医療における治験デザインの結論:科学的合理性と柔軟な設計が承認の鍵

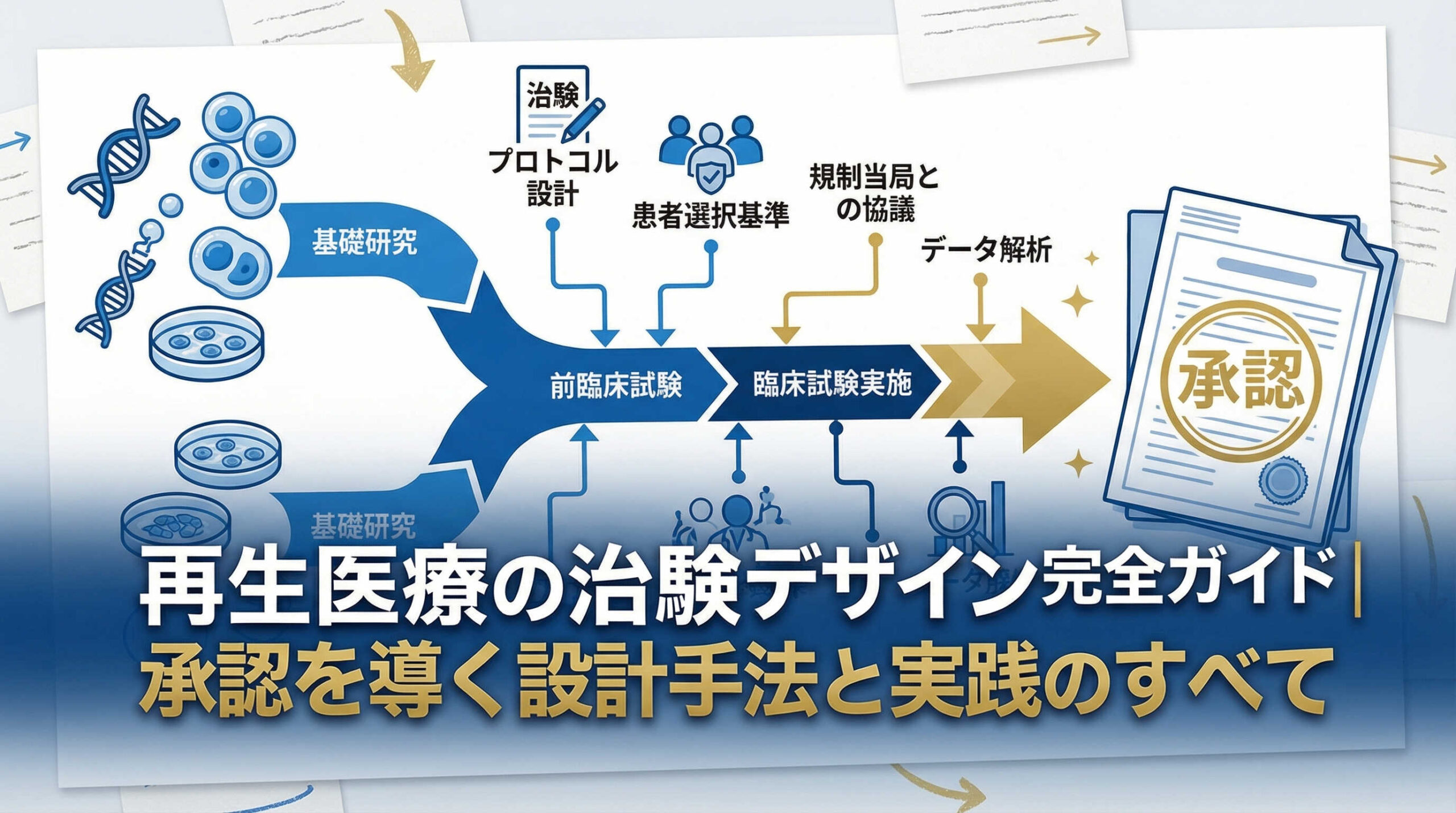
再生医療等製品の開発において、治験デザインの策定は承認取得の成否を分ける最も重要なプロセスの一つです。
従来の医薬品開発で標準とされる大規模なランダム化比較試験(RCT)は、科学的根拠のレベルが高い一方で、再生医療の現場では必ずしも現実的ではないケースが多々あります。
ここでは、再生医療における治験デザインの核心となる考え方について、3つの視点から解説いたします。
従来のRCT(ランダム化比較試験)に固執しない最適なデザインの選択
これまでの医薬品開発では、二重盲検ランダム化比較試験(RCT)が有効性を証明するための最良の方法とされてきました。しかし、再生医療においては、対象疾患が希少であったり、倫理的な観点からプラセボ群を置くことが難しかったりと、RCTの実施が困難な場面が多く見受けられます。
そのため、必ずしもRCTに固執する必要はありません。重要なのは、その試験デザインが「科学的に合理的であるか」そして「実施可能であるか(Feasibility)」という点です。製品の特性や対象疾患の状況を鑑み、単群試験や外部対照試験など、代替となるデザインを選択する論理的な根拠を明確にすることが求められます。
条件付き期限付承認制度の活用を視野に入れた計画策定
日本には、再生医療等製品に対する独自の早期承認制度である「条件付き期限付承認制度」が存在します。これは、有効性が推定され、安全性が確認されれば、特別に早期に承認を与え、市販後に有効性・安全性のデータを収集・確認することを認めるものです。
治験デザインを検討する際は、確定的な検証的試験(第III相試験)で完全な優越性を示すことだけを目指すのではなく、この制度の活用も視野に入れた戦略的な計画策定が有効です。限られた症例数でどのように「有効性の推定」を行うか、そのロジックを組み立てることが開発期間の短縮につながります。
PMDA(医薬品医療機器総合機構)との早期相談による合意形成の重要性
どれほど理論的に優れた治験デザインであっても、規制当局であるPMDAの合意が得られなければ開発は前に進みません。特に再生医療のような新規性の高い分野では、ガイドラインに沿うだけでなく、個別の品目ごとに柔軟な判断がなされることが一般的です。
したがって、プロトコルを完全に固めてから相談に行くのではなく、開発の早期段階からPMDAの治験相談を活用し、試験デザインの骨子について認識をすり合わせておくことが極めて重要です。規制当局を「審査する側」と捉えるのではなく、共に最適な評価方法を模索するパートナーとして対話を重ねる姿勢が、スムーズな承認への近道となります。
再生医療等製品の開発において標準的な治験デザインが困難な理由

低分子医薬品や抗体医薬品の開発と比較して、再生医療等製品には特有の難しさがあります。それは単に技術的な問題だけでなく、対象となる患者層や倫理的な制約など、複合的な要因が絡み合っているためです。
なぜ再生医療では「教科書通り」の治験デザインが通用しないのか、その背景にある主要な理由を整理して理解しておきましょう。
希少疾患対象による症例数確保の限界と統計的検出力の問題
再生医療が対象とする疾患は、難治性疾患や希少疾患であることが多く、そもそも対象となる患者数が極めて少ないという課題があります。
統計学的な観点から言えば、サンプルサイズ(症例数)が少なくなればなるほど、治療効果の有意差を検出する力(検出力)は低下します。数百人規模のデータを必要とする通常の統計解析手法をそのまま適用しようとすると、現実的に集積不可能な症例数が必要となり、治験自体が成立しなくなるリスクがあります。そのため、小規模なデータセットでも科学的な推論が可能となるような工夫が不可欠なのです。
外科的手技を伴う場合のシャム(偽)手術と倫理的課題
多くの再生医療等製品は、移植手術やカテーテル留置といった外科的な手技を伴います。
薬の飲み薬であれば、有効成分を含まない偽薬(プラセボ)を用いることで容易に盲検化が可能ですが、外科的処置を伴う場合、比較対照群に対して皮膚切開のみを行う「シャム(偽)手術」を実施することは、患者への身体的侵襲やリスクを考慮すると倫理的に極めて高いハードルがあります。このため、実薬群と対照群を厳密に比較するデザインを組むことが難しく、評価バイアスをどのように排除するかが大きな課題となります。
細胞製品特有の不均一性と品質管理の難しさ
化学合成された医薬品とは異なり、細胞加工製品は「生きている細胞」そのものを扱います。ドナーの個体差や培養条件の微細な変化により、最終製品の品質にどうしても一定のばらつき(不均一性)が生じてしまいます。
この製品ごとの不均一性は、臨床試験における治療効果のばらつきとして現れる可能性があります。治験デザインにおいては、このばらつきを許容範囲として捉えるのか、あるいは特定の品質基準で厳格に層別化するのか、品質管理(CMC)と臨床評価の連携がこれまで以上に重要になります。
治療効果の不可逆性と長期的な安全性追跡の必要性
一度体内に投与・移植された細胞等は、医薬品のように投与を中止すれば消失するものではなく、長期間にわたって体内に生着し、作用し続ける可能性があります。これは治療効果の持続というメリットである反面、予期せぬ副作用が起きた場合に対処が難しいというリスクも孕んでいます。
また、腫瘍化などのリスク評価には数年単位の観察が必要となることもあります。治験デザインにおいては、有効性の評価期間だけでなく、長期的な安全性をどのように追跡(フォローアップ)するかという計画もセットで検討しなければなりません。
再生医療で検討すべき具体的な治験デザインの種類

前述のような課題を乗り越えるために、再生医療の治験では従来の枠にとらわれない多様なデザインが検討されています。
ここでは、実際にPMDAとの相談や承認事例で見られる、再生医療に適した具体的な治験デザインの種類をご紹介します。自社の開発品目がどのパターンに適しているか、検討の材料にしてみてください。
単群試験(シングルアーム)の適用可能性と要件
対照群を置かず、治験薬を投与する単一のグループのみで実施する試験デザインです。希少疾患などで症例数が極端に少ない場合や、過去の治療成績から効果が明らかに見込めない疾患に対して劇的な効果が期待される場合に選択肢となります。
このデザインを採用するためには、事前に「閾値(これ以上の効果があれば有効とする基準)」を明確に設定し、その根拠を過去の文献や診療データから論理的に説明できることが必須要件となります。バイアスが入りやすいため、客観的な評価項目の設定が鍵となります。
外部対照(ヒストリカルコントロール)を用いた比較試験の設計
治験の中で対照群を設ける代わりに、過去の診療録(カルテ)データや疾患レジストリ(登録制度)のデータを「外部対照」として利用し、治験群と比較する方法です。
新たに患者を対照群に割り付ける必要がないため、症例登録の負担を軽減できるメリットがあります。ただし、過去のデータと今回の治験データの背景因子(重症度、併用薬、治療環境など)が異なると正確な比較ができないため、厳密なマッチングや統計的な調整が必要となります。データの質と均質性が担保できるかが成功の分かれ目です。
プラセボ対照を置かない非盲検ランダム化比較試験
シャム手術が困難な場合など、医師も患者もどの治療を受けたかを知っている状態(非盲検)で、標準治療群などと比較を行うランダム化比較試験です。
盲検化されていないため、主観的な評価(痛みや気分の改善など)にはバイアスがかかりやすくなります。これを防ぐため、治療を行う医師とは別に、治療内容を知らされていない第三者が評価を行う「評価者盲検(PROBE法)」を取り入れるなどの工夫が求められます。客観的な画像データや検査値を主要評価項目に据えることも重要です。
クロスオーバー試験やアダプティブデザインの活用
慢性疾患で症状が安定している場合、一人の患者に対して順序を変えて実薬とプラセボの両方を投与する「クロスオーバー試験」も検討の余地があります。少ない症例数で効率的にデータを収集できますが、再生医療の場合は持ち越し効果(前の治療効果が残ること)の影響が大きいため、適用は慎重に判断する必要があります。
また、治験の途中で得られたデータに基づいて、症例数や評価項目を柔軟に変更する「アダプティブデザイン」も、不確実性の高い再生医療開発において有効な戦略となり得ます。
製造販売後調査(全例調査)を前提とした承認申請データの構築
条件付き期限付承認を目指す場合、治験段階では「有効性の推定」にとどめ、承認後の全例調査(製造販売後調査)で詳細なデータを収集するというパッケージで計画を立てることもあります。
この場合、治験プロトコルの段階から、市販後にどのようなデータを、どの程度の期間、どのような体制で収集するのかを具体的に設計しておく必要があります。治験と市販後調査を一連の流れとして捉え、切れ目のないデータ収集体制(レジストリの活用など)を構築することが、当局の信頼獲得につながります。
治験の成功確率を高める主要評価項目(エンドポイント)の設定

治験が成功するかどうか、つまり「有効性あり」と判定されるかどうかは、主要評価項目(プライマリーエンドポイント)を何に設定するかで大きく変わります。
再生医療においては、単に検査値が改善するだけでなく、患者さんの生活の質がどう変わったかという視点も重要視されています。ここでは、適切な評価項目の選び方について解説します。
臨床的意義(Clinical Relevance)に基づいた評価指標の選定
評価項目を選ぶ際、最も大切なのは「臨床的意義(Clinical Relevance)」です。統計的に有意差が出たとしても、それが実際の患者さんにとって「意味のある改善」でなければ、治療薬としての価値は認められません。
例えば、歩行速度が0.1秒速くなったとしても、患者さんの日常生活が変わらなければ意義は薄いかもしれません。「自力でトイレに行けるようになる」「痛みがなく眠れるようになる」など、具体的な臨床上のメリットを反映した指標を選定することが、承認審査においても説得力を持ちます。
サロゲートエンドポイント(代替指標)の活用と妥当性の説明
真の評価項目(生存期間や発症予防など)の観察に長い年月を要する場合、それと相関関係にある代替指標(サロゲートエンドポイント)を用いることがあります。再生医療では、画像所見や特定のバイオマーカーなどがこれに該当します。
サロゲートエンドポイントを主要評価項目とする場合、その指標が真の臨床効果をどれだけ正確に予測できるか、過去の文献やデータを用いて科学的に説明できなければなりません。PMDAとの相談でも、この「妥当性」の説明が重要な論点となります。
構造的改善(画像評価等)と機能的改善のバランス
再生医療では、組織の修復や再生を目指すため、MRIやCTなどの画像評価(構造的改善)が重視されがちです。しかし、画像上で組織が再生していても、実際の臓器機能や運動機能(機能的改善)が伴っていなければ治療としての意味をなしません。
治験デザインにおいては、構造的な改善(形態)と機能的な改善(働き)のどちらを主要評価項目にするか、あるいは両者をどう組み合わせるか、対象疾患の病態生理に合わせて慎重にバランスを取る必要があります。
患者報告アウトカム(PRO)の導入と評価方法
近年、規制当局が特に注目しているのが、患者自身が自分の健康状態について報告する「患者報告アウトカム(PRO: Patient Reported Outcome)」です。痛み、疲労感、身体機能、QOL(生活の質)など、医師の評価だけでは捉えきれない患者主観のメリットを評価します。
主要評価項目とまではいかなくとも、副次評価項目としてPROを適切に組み込むことで、製品の多面的な価値を示すことができます。評価には、妥当性が検証された標準的な質問票を用いることが一般的です。
小規模データにおける統計解析手法とバイアス制御

再生医療の治験では、どうしても症例数が限られるため、大規模データのような正規分布を前提とした解析が難しいことがあります。
少ないデータからいかに信頼性の高い結果を導き出し、バイアス(偏り)を最小限に抑えるか。ここでは、統計解析の専門的な視点から、小規模データにおけるアプローチ方法を解説します。
限られた症例数での統計的有意差の検証方法
症例数が少ない場合、通常のt検定などでは有意差が出にくいことがあります。このような場合、ノンパラメトリック検定の使用や、ベイズ統計のアプローチを検討することもあります。
しかし、統計的な有意差(p値0.05未満など)だけにこだわりすぎると、本質を見誤る可能性があります。PMDAの審査においても、p値だけでなく、効果の大きさ(推定値の点推定値)や信頼区間、そして個々の症例の臨床経過を詳細に検討する「記述統計的」な評価が重視される傾向にあります。数字の背後にある臨床的な意味を丁寧に解釈する姿勢が必要です。
共変量調整や傾向スコアを用いた外部対照との比較
外部対照を用いる場合、治験群と外部対照群の間には背景因子のズレが生じがちです。これを調整するために、重回帰分析などの共変量調整や、傾向スコア(プロペンシティスコア)を用いたマッチングなどの手法が用いられます。
これにより、年齢、性別、重症度などの条件を統計的に揃え、擬似的にランダム化に近い状態で比較を行うことが可能になります。ただし、これらの手法を適用するためには、外部対照データの質が高く、必要な項目が欠損なく収集されていることが前提となります。
欠測データの取り扱いと感度分析の重要性
長期の追跡が必要な再生医療の治験では、患者の転居や脱落によりデータの一部が欠測する(取れなくなる)リスクがあります。欠測データを「なかったこと」にして解析から除外すると、結果に大きなバイアスが生じます。
LOCF(直近の値を代入する方法)やMMRM(混合効果モデル)など、適切な統計手法を用いて欠測データを処理することが重要です。また、異なる前提条件で解析を行っても結果が変わらないことを確認する「感度分析」を実施し、結果の頑健性(Robustness)を示すことが審査での信頼性向上につながります。
中間解析の実施とサンプルサイズ再設計の検討
治験の途中でデータを解析する「中間解析」を実施することで、想定よりも効果が高い場合に早期に治験を終了したり、逆に効果が見込めない場合に無益な治験を中止したりする判断が可能になります。
また、当初の設定よりもばらつきが大きかった場合に、統計的な検出力を保つためにサンプルサイズ(症例数)を再設計(追加)する手法もあります。これらは事前にプロトコルに規定しておく必要がありますが、開発リスクをコントロールする有効な手段となります。
PMDAの治験相談をスムーズに進めるためのプロトコル策定準備

治験デザインの骨子が固まったら、次はいよいよPMDAとの対面助言(治験相談)に向けた準備です。
相談の場で建設的な議論を行い、こちらの提案を受け入れてもらうためには、単に「やりたいこと」を伝えるだけでは不十分です。ここでは、PMDA相談をスムーズに進め、合意形成を図るための実践的な準備ポイントをお伝えします。
治験デザインの科学的根拠を示すための資料作成
PMDAの担当者が最も気にするのは、「なぜそのデザインでなければならないのか」という必然性です。「症例が集まらないから」という消極的な理由だけでなく、疾患の特性や既存治療の限界、製品の作用機序などを踏まえた積極的な科学的根拠(ラショナル)を示す必要があります。
国内外のガイドライン、学術論文、専門家の意見書などを資料として整理し、「このデザインこそが、本製品の有効性と安全性を評価するのに最適である」というストーリーを論理的に構築して提示しましょう。
先行品目の審査報告書から読み解く規制当局の判断基準
PMDAのウェブサイトで公開されている、過去に承認された再生医療等製品の「審査報告書」は、情報の宝庫です。
類似の製品や対象疾患が近い品目が、どのような治験デザインで承認されたのか、またはどのような点で指摘を受け、どう回答してクリアしたのかが詳細に記録されています。これらを読み解くことで、規制当局が重視しているポイントや判断基準(相場観)を把握することができます。先人の事例を徹底的に分析し、自社の戦略に活かしましょう。
アカデミア主導臨床研究データの有効活用(医師主導治験への移行)
再生医療の開発では、大学や研究機関で行われた「医師主導臨床研究」のデータを基礎として、企業治験へ移行するケースが多くあります。
このアカデミアデータを企業の承認申請データとして有効活用できるかは、データの質(GCP準拠性や信頼性保証)に大きく依存します。早期からアカデミアと連携し、将来の承認申請を見据えたプロトコルやデータ管理体制を構築しておくことで、企業治験の期間短縮やコスト削減につなげることが可能です。
まとめ

再生医療における治験デザインは、従来の医薬品開発の枠組みを超えた柔軟性と科学的な厳密さの両立が求められる高度な領域です。
希少疾患や細胞製品特有の課題に対して、単群試験や外部対照の活用、条件付き期限付承認制度の利用など、様々なアプローチが存在します。しかし、どの手法を選択するにしても、最も重要なのは「患者さんにとっての利益」と「科学的な妥当性」を論理的に結びつけることです。
PMDAとの早期相談を通じて規制当局と認識を共有し、先行事例を参考にしながら、貴社の製品に最適な「勝ち筋」となる治験デザインを設計していってください。
治験デザインについてよくある質問

最後に、再生医療の治験デザインを検討する際によく寄せられる質問をまとめました。
実務を進める上での疑問点として、ぜひ参考にしてください。
- 治験デザインの策定はどの段階から開始すべきですか?
- 海外データ(ブリッジング試験)は再生医療でも活用できますか?
- 外部対照として利用できるレジストリデータにはどのような要件がありますか?
治験デザインの策定はどの段階から開始すべきですか?
非臨床試験(動物実験など)でPOC(概念実証)が得られる前後から検討を開始するのが理想的です。
最終的なプロトコル確定は非臨床試験の結果を踏まえてからになりますが、早い段階から「どのような患者層を狙うか」「エンドポイントを何にするか」という骨子を考えておくことで、非臨床試験のデザイン自体も、臨床を見据えたより有用なものに調整できるからです。また、PMDA相談の予約枠確保などのスケジュール管理の観点からも、早期着手が望ましいでしょう。
海外データ(ブリッジング試験)は再生医療でも活用できますか?
活用は可能ですが、慎重な検討が必要です。
ICH E5ガイドライン(民族的要因に関するガイドライン)の考え方は再生医療にも適用されますが、細胞製品の場合、人種による遺伝的背景の違いや、医療環境(標準治療)の違いが結果に与える影響が低分子医薬品よりも複雑になる可能性があります。海外データを日本での承認申請に用いる場合は、日本人集団でも同様の有効性・安全性が期待できることを示すための追加データや、詳細な考察が求められます。
外部対照として利用できるレジストリデータにはどのような要件がありますか?
外部対照として利用するためには、データの「信頼性」と「適合性」が重要です。
具体的には、データが網羅的に収集されていること、入力基準が統一されていること、監査証跡が残っていることなど、ある程度の質の担保(GCPに準ずる信頼性)が求められます。また、治験の対象患者とレジストリの患者の背景因子が比較可能であることも必須です。既存のレジストリをそのまま使えるケースは稀で、多くの場合、事前のデータクリーニングや補完調査が必要になります。