再生医療等製品の開発において、製造販売承認の取得は極めて重要なマイルストーンですが、ビジネスとしての真の成功を決定づけるのは、その後に続く「商用生産体制の確立」です。多くの企業が、治験薬製造から商用製造への移行フェーズにおいて、製造コストの高騰や品質保証の厳格化といった予期せぬ壁に直面しています。
「上市後の製造計画」を早期に策定し、開発段階から商用化を見据えたCMC(Chemistry, Manufacturing and Control)戦略を練り上げることは、これらのリスクを最小化し、事業の収益性を確保するために不可欠です。本記事では、再生医療業界のCMC担当者や事業開発責任者の方々に向けて、上市後に直面する製造上の課題と、その解決に向けた具体的かつ実践的なロードマップを解説します。安定供給とコスト最適化を両立させるためのヒントとして、ぜひお役立てください。
再生医療における上市後の製造計画とは?商用化成功の鍵は「ギャップの解消」

再生医療等製品の上市に向けた準備において、最も重要な視点は「治験薬製造」と「商用製造」の間にあるギャップを正確に認識し、それを埋めることです。臨床試験では有効性と安全性の証明が最優先されますが、上市後はそれに加えて「恒常的な安定供給」と「経済合理性」が強く求められます。このフェーズでの判断ミスは、後の事業収益に長期的な影響を及ぼすため、慎重な計画策定が必要です。
治験薬製造と商用製造における品質保証レベルの決定的な違い
治験薬製造と商用製造では、求められる品質保証(QA)のレベルと視点が異なります。治験段階では、限られたバッチ数で特定の患者群に対する安全性が確保されていれば許容される側面がありますが、商用生産では不特定多数の患者へ、長期間にわたり均一な品質の製品を供給し続ける義務が生じます。
具体的には、GCTP(Good Gene, Cellular, and Tissue-based Products Manufacturing Practice)省令に基づいた、より堅牢な製造管理および品質管理体制が求められます。逸脱管理や変更管理のプロセスもより厳格になり、製造プロセスの一貫性(Consistency)を科学的データに基づいて常に証明し続ける体制が必要となるのです。
承認取得後を見据えたCMC戦略が事業収益性に与える影響
承認取得後のCMC戦略は、製品の原価率、ひいては事業の収益性に直結します。再生医療等製品は原材料費や製造に関わる人件費が高額になりがちであり、開発早期から商用スケールでのコスト構造(CoGs:Cost of Goods Sold)を意識したプロセス設計を行わなければ、薬価に見合わない高コスト体質に陥るリスクがあります。
例えば、使用する培地や試薬のグレード、製造プロセスの自動化レベル、バッチサイズの最適化などは、後の変更が困難な要素です。これらを商用化の視点で早期に最適化しておくことが、上市後の利益率を最大化するための重要な戦略となります。
早期に「製造販売承認書」と「実製造」の整合性を図る重要性
上市後に頻発するトラブルの一つに、承認申請書(製造販売承認書)の記載内容と、実際の製造現場での手順に乖離が生じる「承認書齟齬」があります。これは重大なコンプライアンス違反となり、製品回収や業務停止命令につながる恐れがあります。
このような事態を防ぐためには、申請段階から製造現場の実態を正確に反映させた記述を行うとともに、承認取得後も定期的に自己点検を行い、整合性を確認することが不可欠です。製造部門と薬事部門が密に連携し、現場の些細な変更も適切に管理・反映できる体制を早期に構築することが、コンプライアンスリスクの低減につながります。
再生医療等製品の商用化で直面する製造上の主要課題(Reason)

再生医療等製品が商用化フェーズに入ると、研究開発段階では顕在化しなかった製造上の課題が浮き彫りになります。これらは製品の安定供給を脅かすだけでなく、ビジネスモデルそのものを揺るがす要因となり得ます。ここでは、多くの企業が直面する主要な4つの課題について、その背景と理由を掘り下げます。
製造コスト(CoGs)の高騰と薬価収載を見据えた原価低減の難しさ
再生医療製品、特に自家細胞製品においては、製造原価(CoGs)の高騰が深刻な課題です。高額なサイトカインや培地、特殊な使い捨て部材の使用に加え、高度なクリーンルーム維持費が原価を押し上げます。
一方で、薬価算定においては製造原価がそのまま反映されるとは限らず、厳しい薬価が設定されるケースも少なくありません。上市後に原価低減を図ろうとしても、原材料の変更には同等性評価や一部変更承認申請(一変)が必要となり、多大な時間とコストがかかります。したがって、開発段階から「目標とする原価」を設定し、それを達成できる製造プロセスを構築しておくことが求められます。
手作業依存からの脱却と製造プロセスの自動化・効率化
多くの治験薬製造は、熟練した技術者の手技に依存するマニュアル操作で行われています。しかし、商用生産で数千、数万の患者に製品を届けるためには、手作業による「ばらつき」を排除し、生産性を向上させる必要があります。
手作業への依存は、ヒューマンエラーのリスクを高めるだけでなく、スケールアウト(並列化)時の要員確保や教育コストの増大を招きます。閉鎖系自動培養装置の導入やロボティクス技術の活用によるプロセスの自動化・効率化は、品質の安定化とコスト削減を両立させるための必須要件と言えるでしょう。
GCTP省令に準拠した厳格な製造管理・品質管理体制(QMS)の構築
再生医療等製品の製造管理および品質管理は、GCTP省令に準拠する必要があります。これは医薬品のGMPと比較しても、製品の特性(不均一性や無菌性の確保など)に応じた、より高度で柔軟な管理が求められるものです。
特に、原材料の受け入れから製造、出荷判定に至るまでの全工程において、無菌操作の保証や交叉汚染(クロスコンタミネーション)の防止策を講じることは容易ではありません。また、これらの管理状況を記録・保存し、常に査察に対応できる状態を維持するためのQMS(品質マネジメントシステム)構築には、専門的な知識と多くのリソースが必要となります。
コールドチェーンを含む物流管理とトレーサビリティの確保
細胞製品の多くは、凍結状態や特定の温度帯での厳格な輸送管理が求められます。製造拠点から医療機関までの「コールドチェーン」を確立し、温度逸脱がないことを証明するトレーサビリティの確保は、製品の有効性を担保する上で生命線となります。
輸送中の振動や温度変化が細胞品質に与える影響を検証し、適切な輸送容器やデータロガーを選定する必要があります。また、物流業者の選定や教育、緊急時の対応フロー策定など、製造所の外側まで含めた品質保証体制を敷くことが、商用化における大きな課題となります。
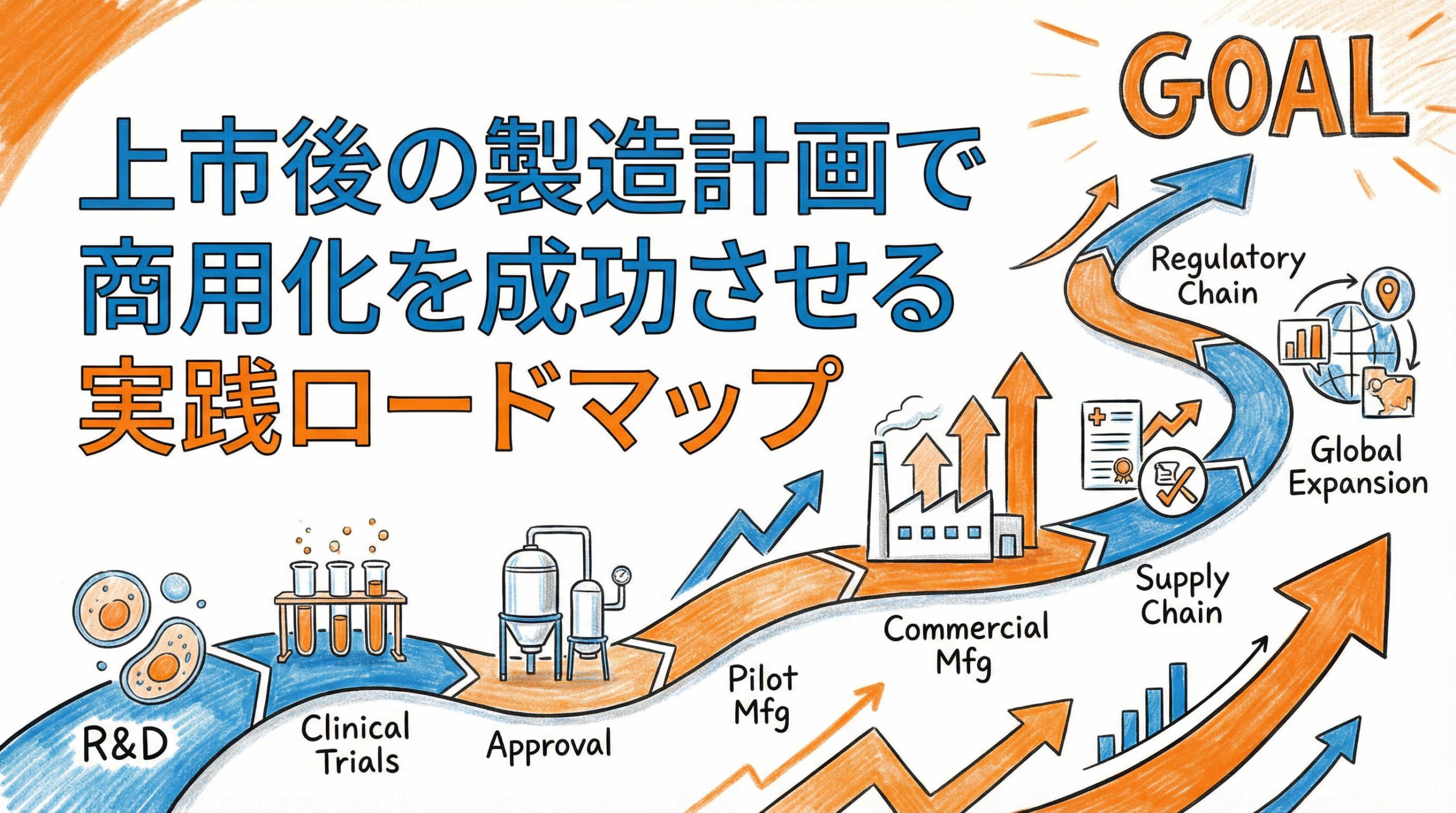
上市に向けた製造計画の具体的な策定ロードマップ(Example)

上市後のトラブルを未然に防ぎ、スムーズな商用生産を開始するためには、臨床試験の進行に合わせて段階的に製造計画を具体化していく必要があります。ここでは、臨床試験後期から承認取得後に至るまでの、理想的な製造計画策定のロードマップを提示します。
臨床試験後期(Phase III)における製造プロセスの固定化(Process Freezing)
臨床試験の第III相(Phase III)に入る段階で、製造プロセスは商用生産と同一のものに固定化(Process Freezing)されていることが理想的です。この段階でプロセスが固まっていない場合、臨床試験データと商用製品との同等性を証明することが困難になり、承認審査の遅延や追加試験の原因となります。
具体的には、重要工程パラメータ(CPP)や重要品質特性(CQA)を特定し、管理幅を設定します。将来的なスケールアップを見越している場合でも、基本となる培養条件や使用する原材料はこの時点で確定させ、変更リスクを最小限に抑える戦略が重要です。
商用生産を想定したプロセスバリデーション(PV)の実施計画
商用生産の開始前には、設定された製造プロセスが恒常的に品質適合製品を製造できることを科学的に証明する「プロセスバリデーション(PV)」を実施します。一般的には、実生産スケールで連続3ロット以上の製造を行い、すべての品質基準を満たすことを確認します。
PVの実施計画書には、検証項目、判定基準、サンプリング計画などを詳細に規定します。再生医療製品は原材料のばらつきが大きいため、その変動を許容できる堅牢なプロセスであることを示すデータ取得が鍵となります。このPVデータは、承認申請資料の重要な根拠となります。
製造販売承認申請(J-NDA)に向けたモジュール3(品質文書)の作成
製造販売承認申請(J-NDA)におけるCTDモジュール3(品質に関する文書)の作成は、製造計画の集大成とも言える作業です。ここでは、開発の経緯、製造方法、特性解析、規格及び試験方法、安定性試験などのデータを論理的に構成し、品質の安全性を主張します。
特に、QbD(Quality by Design)アプローチに基づき、リスク評価を経てどのように管理戦略(Control Strategy)を決定したかを明確に記述することが求められます。審査当局からの照会事項(PMDA相談などでの指摘)を想定し、データの信頼性と網羅性を高めておくことが、早期承認への近道です。
承認取得後の定期的なバリデーションと年次レビュー体制の整備
承認取得はゴールではなく、継続的な品質保証のスタートです。上市後は、定期的なバリデーション(再バリデーション)や、製品品質照査(PQR:Product Quality Review)を通じて、製造プロセスが管理された状態にあるかを年次で評価します。
得られたデータをトレンド分析し、品質低下の兆候があれば予防措置を講じます。また、新たな知見や技術進歩に合わせて継続的な改善を行うことも重要ですが、その際は変更管理手順に従い、承認内容との整合性を常に維持する体制整備が必要です。
製造プロセスの変更と同等性・同質性の評価(Comparability)

商用化に伴い、製造スケールの拡大や場所の変更、原材料の切り替えなど、プロセスの変更が必要になる場面は多々あります。このとき重要になるのが、変更前後で製品の品質が同等であることを証明する「同等性・同質性(Comparability)」の評価です。適切な評価戦略なしに変更を行うと、これまでの臨床データを無効にしてしまうリスクがあります。
スケールアップ・スケールアウトに伴う品質変動リスクの管理
研究レベルのフラスコ培養から、商用レベルのバイオリアクターへスケールアップする際、細胞にかかる物理的ストレス(せん断力など)や酸素供給効率が変化し、細胞の特性が変わってしまうリスクがあります。あるいは、自家細胞製品のようにスケールアウト(並列化)する場合でも、操作間の差が生じやすくなります。
これらのリスクを管理するためには、変更前後の製品で、表面マーカー、分泌タンパク質、遺伝子発現プロファイルなどの特性解析(Characterization)を詳細に行い、品質特性に有意な差がないことをデータで示す必要があります。
製造場所変更や原材料変更時における同等性評価の手順
製造拠点を変更する場合や、重要原材料(培地や血清など)のサプライヤーを変更する場合、同等性評価は避けて通れません。評価手順としては、まずリスクアセスメントを行い、変更が品質に与える影響の度合いを予測します。
その上で、in vitroでの特性解析試験に加え、必要に応じてin vivoでの非臨床試験、場合によっては小規模な臨床試験が求められることもあります。特に生物由来原料の変更は影響が予測しづらいため、複数のロットを用いた比較試験を行い、統計的な同等性を慎重に検証するプロセスが求められます。
承認事項一部変更承認申請(一変)が必要となるケースと回避策
製造プロセスの変更が、製品の品質・有効性・安全性に影響を与える可能性がある場合、「承認事項一部変更承認申請(一変申請)」が必要となります。これには審査期間が必要であり、承認されるまで新しいプロセスでの製造・出荷はできません。
一方で、品質に影響を与えない軽微な変更であれば、「軽微変更届」で済む場合もあります。一変申請は事業計画に大きなタイムラグを生じさせるため、開発段階でプロセスの変動幅(デザインスペース)を広く設定しておくなど、可能な限り一変申請を回避、あるいは回数を減らすための戦略的な申請資料作成が重要です。
安定供給とコスト最適化を実現する製造体制の選択肢

再生医療製品を市場へ安定的に供給し、かつコスト競争力を維持するためには、どのような製造体制を構築するかが経営上の重要な意思決定となります。自社ですべてを賄うのか、外部パートナーを活用するのか。ここでは、製造体制の選択肢とそのメリット・デメリット、リスク管理について解説します。
自社製造(内製化)とCDMO活用のメリット・デメリット比較
自社製造(内製化)は、製造ノウハウを社内に蓄積でき、トラブル時の迅速な対応や細かなプロセス改善が容易である反面、初期投資(設備建設費)や維持管理費(固定費)が重くのしかかります。
一方、CDMO(医薬品開発製造受託機関)の活用は、初期投資を抑え、製造コストを変動費化できる点が最大のメリットです。また、CDMOが持つ専門的な規制対応ノウハウを活用できます。しかし、技術移転に時間を要する点や、製造委託先のスケジュールに依存するリスク、自社にノウハウが残りにくいというデメリットも考慮し、フェーズに応じた使い分けが重要です。
デュアルサイト(複数製造拠点)によるBCP対策とリスク分散
再生医療製品は、患者の生命に直結する場合が多く、供給停止が許されないケースがあります。自然災害やパンデミック、設備の故障などの不測の事態に備え、BCP(事業継続計画)の一環として「デュアルサイト(複数製造拠点)」体制を構築することが推奨されます。
例えば、メインの製造を自社工場で行い、バックアップとしてCDMOを確保しておく、あるいは地理的に離れた2つの拠点で製造認定を取得しておくなどの対策です。これにより、片方の拠点が停止しても供給を継続できるリスク分散が可能となります。
サプライチェーンマネジメント(SCM)における重要原材料のセカンドソース確保
製造体制だけでなく、原材料の供給網(サプライチェーン)のリスク管理も極めて重要です。特定の国や特定のサプライヤー1社に依存している原材料(シングルソース)は、地政学リスクや企業の倒産などで供給が途絶える恐れがあります。
特に、培地やサイトカイン、特殊なプラスチック製品などの重要原材料については、早期に代替品を評価し、セカンドソース(第二の供給元)を確保しておくことが安定供給の要です。承認書に複数の原材料を記載しておくことで、緊急時の切り替えをスムーズに行うことができます。
まとめ

再生医療における上市後の製造計画は、単なる工場の稼働計画ではなく、事業の成功を左右する経営戦略そのものです。治験薬製造と商用製造のギャップを早期に認識し、GCTP準拠の品質保証体制、コスト競争力のあるプロセス、そして堅牢なサプライチェーンを構築することが求められます。
臨床試験の進行と並行して、商用化を見据えたCMC戦略を緻密に練り上げ、プロセスバリデーションや同等性評価を計画的に進めることで、手戻りのないスムーズな上市が可能となります。リスクを先回りして管理し、患者様へ安定的に製品を届ける体制を築くことこそが、再生医療ビジネスにおける最大の競争優位性となるでしょう。
上市後の製造計画についてよくある質問

上市後の製造計画に関して、再生医療事業の担当者から頻繁に寄せられる質問をまとめました。商用化に向けた疑問の解消にお役立てください。
Q1. 治験薬製造と商用製造で、最も意識すべき違いは何ですか?
- A. 最大の違いは「恒常性」と「コスト意識」です。治験では有効性証明が優先されますが、商用ではGCTPに基づき、常に均一な品質を保証し続ける体制と、事業として成立する原価管理が必須となります。
Q2. 製造コスト(CoGs)を下げるための具体的なポイントは?
- A. 手作業の自動化による人件費削減、安価で高品質な代替原材料への切り替え、スケールアップによるバッチあたりの収量増加が主要なポイントです。これらを開発早期に検討することが重要です。
Q3. 製造場所を変更する際、同等性評価で失敗しないコツは?
- A. 変更前のプロセス理解(デザインスペースの把握)を深めておくことです。また、リスクアセスメントを入念に行い、特性解析だけでなく、必要に応じた機能試験を組み込むなど、論理的な評価計画を立てることが重要です。
Q4. CDMOを選定する際、どのような基準で選ぶべきですか?
- A. 再生医療分野での実績はもちろん、自社の製品特性(細胞種や培養方法)に近い経験があるか、技術移転の体制が整っているか、そして規制当局との折衝経験が豊富かどうかが重要な選定基準となります。
Q5. 上市後に製造プロセスを変更する場合、「一変申請」の判断基準は?
- A. 変更が製品の「品質、有効性、安全性」に影響を与えるかどうかが基準です。影響がある場合は一変申請、軽微な場合は軽微変更届となります。判断に迷う場合は、PMDAの事前面談などを活用することをお勧めします。